Shared memory parallelism: multithreading & multiprocessing
Videos
Videos of this topic may be available from one of our kickstart course playlists: 2025 summer, 2025 winter, 2024, 2023, 2022 Summer, 2022 February, 2021 Summer, 2021 February. Or see computing explained with HPC kitchen.
Abstract
Verify that your program can use multiple CPUs.
Use
--cpus-per-task=Cto reserveCCPUs for your job.In your program set the number of used CPUs to match the number of requested CPUs. You can use
$SLURM_CPUS_PER_TASKenvironment variable to get this number dynamically.You must always monitor jobs to make sure they are using all the resources you request (
seff JOBID).If you aren’t fully sure of how to scale up, contact us Research Software Engineers early.
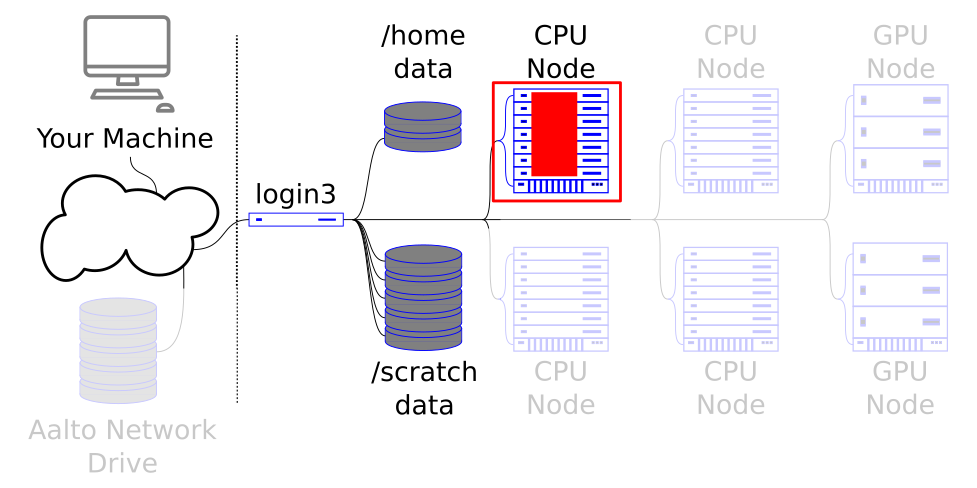
Shared-memory parallelism and multiprocessing lets you scale to the size of one node. For purposes of illustration, the picture isn’t true to life: we call the whole stack one node, but in reality each node is one of the rows.
What is shared memory parallelism?
In shared memory parallelism a program will launch multiple processes or threads so that it can leverage multiple CPUs available in the machine.
Slurm reservations for both methods behave similarly. This document will talk about processes, but everything mentioned would be applicable to threads as well. See section on difference between processes and threads for more information on who proceseses and threads differ.
Communication between processes happens via shared memory. This means that all processes need to run on the same machine.

Depending on a program, you might have multiple processes (Matlab parallel pool, R parallel-library, Python multiprocessing) or have multiple threads (OpenMP threads of BLAS libraries that R/numpy use).
Running a typical multiprocess program
Special cases and common pitfalls
Monitoring CPU utilization for parallel programs
You can use seff JOBID to see what percent of available CPUs and RAM was
utilized. Example output is given below:
$ seff 60985042
Job ID: 60985042
Cluster: triton
User/Group: tuomiss1/tuomiss1
State: COMPLETED (exit code 0)
Nodes: 1
Cores per node: 2
CPU Utilized: 00:00:29
CPU Efficiency: 90.62% of 00:00:32 core-walltime
Job Wall-clock time: 00:00:16
Memory Utilized: 1.59 MB
Memory Efficiency: 0.08% of 2.00 GB
If your processor usage is far below 100%, your code may not be working correctly. If your memory usage is far below 100% or above 100%, you might have a problem with your RAM requirements. You should set the RAM limit to be a bit above the RAM that you have utilized.
You can also monitor individual job steps by calling seff with the syntax
seff JOBID.JOBSTEP.
Important
When making job reservations it is important to distinguish
between requirements for the whole job (such as --mem) and
requirements for each individual task/cpu (such as --mem-per-cpu).
E.g. requesting --mem-per-cpu=2G with --ntasks=2 and --cpus-per-task=4
will create a total memory reservation of
(2 tasks)*(4 cpus / task)*(2GB / cpu)=16GB.
Multithreaded vs. multiprocess and double-booking
Processes are individual program executions while threads are basically small work executions within a process. Processes have their own memory allocations and can work independently from the main process. Threads, on the other hand, are smaller parallel executions within the main process. Processes are slower to launch, but due to their independent nature they won’t block each other’s execution as easily as threads can.
Some programs can utilize both multithread and multiprocess parallelism. For example, R has parallel-library for running multiple processes while BLAS libraries that R uses can utilize multiple threads.
When running a program that can parallelise through processes and through threads, you should be careful to check that only one method of parallisation is in effect.
Using both can result in double-booked parallelism where you launch \(N\) processes and each process launches \(N\) threads, which results in \(N^2\) threads. This will usually tank the performance of the code as the CPUs are overbooked.
Often threading is done in a lower level library when they have been
implemented using OpenMP. If you encounter bad performace or you
see a huge number of threads appearing when you use parallel processes
try setting export OMP_NUM_THREADS=1 in your Slurm script.
Over- and undersubscription of CPUs
The number of threads/processes you launch should match the number of requested processors. If you create a lower number, you will not utilize all CPUs. If you launch a larger number, you will oversubscribe the CPUs and the code will run slower as different threads/processes will have to swap in/out of the CPUs and compete for the same resources.
Using threads and processes at the same time can also result in double-booking.
Using $SLURM_CPUS_PER_TASK is the best way of letting your program
know how many CPUs it should use.
See section on using it effectively
for more information.
Using SLURM_CPUS_PER_TASK effectively
The environment variable $SLURM_CPUS_PER_TASK can be utilized in multiple
ways in your scripts. Below are few examples:
Setting a number of workers when
$SLURM_CPUS_PER_TASKis not set:$SLURM_CPUS_PER_TASKis only set when--cpus-per-taskhas been specified. If you want to run the same code in your own machine and in the cluster it might be useful to set a variable likeexport NCORES=${SLURM_CPUS_PER_TASK:-4}and use that in your scripts.Here
$NCORESis set to the number specified by$SLURM_CPUS_PER_TASKif it has been set. Otherwise, it will be set to 4 via Bash’s syntax for setting default values for unset variables.In Python you can use the following for obtaining the environment variable:
import os ncpus=int(os.environ.get("SLURM_CPUS_PER_TASK", 1))
For more information on parallelisation in Python see our Python documentation.
In R you can use the following for obtaining the environment variable:
ncpus <- as.integer(Sys.getenv("SLURM_CPUS_PER_TASK", unset=1))
For more information on parallelisation in R see our R documentation.
In Matlab you can use the following for obtaining the environment variable:
ncpus=str2num(getenv("SLURM_CPUS_PER_TASK"))
For more information on parallelisation in Matlab see our Matlab documentation.
Asking for multiple tasks when code does not use MPI
Normally you should not use --ntasks=n when you want to
run shared memory codes. The number of tasks is only relevant to MPI codes
and by specifying it you might launch multiple copies of your program
that all compete on the reserved CPUs.
Only hybrid parallelization codes should have both --ntasks=n and
--cpus-per-task=C set to be greater than one.
Exercises
The scripts you need for the following exercises can be found in our
hpc-examples, which
we discussed in Using the cluster from a command line (section
Copy your code to the cluster).
You can clone the repository by running
git clone https://github.com/AaltoSciComp/hpc-examples.git. Doing this
creates you a local copy of the repository in your current working
directory. This repository will be used for most of the tutorial exercises.
Shared memory parallelism1: Test scaling
Test scaling of the ngrams code. How many processors should be used? (This is involved enough you might want to follow the steps in the solution).
To get good stats, compute with -n 2 --words (word 2-grams),
and constrain to a single processor archicteure (like the Slurm
option --constraint=skl).
Solution
First, let’s run the scaling tests for character ngrams just for comparison. Note we don’t save the output anywhere so that we don’t measure the time of disk writing, and we constrain to the same type of node (processor architecture) to make sure our results are comparable:
## 100 books
$ srun --constraint=skl -c 1 python3 ngrams/count.py -n 2 /scratch/shareddata/teaching/Gutenberg-Fiction-first100.zip -o /dev/null
$ srun --constraint=skl -c 1 python3 ngrams/count-multi.py -n 2 -t auto /scratch/shareddata/teaching/Gutenberg-Fiction-first100.zip -o /dev/null
$ srun --constraint=skl -c 2 python3 ngrams/count-multi.py -n 2 -t auto /scratch/shareddata/teaching/Gutenberg-Fiction-first100.zip -o /dev/null
$ srun --constraint=skl -c 4 python3 ngrams/count-multi.py -n 2 -t auto /scratch/shareddata/teaching/Gutenberg-Fiction-first100.zip -o /dev/null
$ srun --constraint=skl -c 8 python3 ngrams/count-multi.py -n 2 -t auto /scratch/shareddata/teaching/Gutenberg-Fiction-first100.zip -o /dev/null
Summary table for character ngrams: Speedup is (single core time)/(multi-core time). “Total core time” is (time × number of processors) and is a measure of how much computing resources you actually used:
N processors |
time |
speedup |
total core time |
|---|---|---|---|
single-core version |
24.7 s |
24.7 s |
|
1 |
23.8 s |
1.04 |
23.8 s |
2 |
12.5 s |
1.98 |
25.0 s |
4 |
7.2 s |
3.4 |
28.8 s |
8 |
5.0 s |
4.9 |
40.0 s |
For these, it seems reasonable to go up to 4 cores, since that’s how far you can go with a reasonable speedup and you aren’t wasting too many resources.
Second, let’s compute the same thing but with words (--words). We see
that the speedup is much worse, and it almost doesn’t make sense
to use multi-core at all. This is because word ngrams have much
more output data, and the programs spend more time moving that
data around than computing anything. This is computed for both
100 and 1000 books:
## 1000 books:
$ srun --constraint=skl --time=0-1 --mem=20G -c 1 python3 ngrams/count.py -n 2 --words /scratch/shareddata/teaching/Gutenberg-Fiction-first1000.zip -o /dev/null
$ srun --constraint=skl --time=0-1 --mem=20G -c 1 python3 ngrams/count-multi.py -n 2 --words -t auto /scratch/shareddata/teaching/Gutenberg-Fiction-first1000.zip -o /dev/null
$ srun --constraint=skl --time=0-1 --mem=20G -c 2 python3 ngrams/count-multi.py -n 2 --words -t auto /scratch/shareddata/teaching/Gutenberg-Fiction-first1000.zip -o /dev/null
$ srun --constraint=skl --time=0-1 --mem=20G -c 4 python3 ngrams/count-multi.py -n 2 --words -t auto /scratch/shareddata/teaching/Gutenberg-Fiction-first1000.zip -o /dev/null
$ srun --constraint=skl --time=0-1 --mem=20G -c 8 python3 ngrams/count-multi.py -n 2 --words -t auto /scratch/shareddata/teaching/Gutenberg-Fiction-first1000.zip -o /dev/null
N processors |
time (100 books) |
speedup |
core time used |
time (1000 books) |
speedup |
core time used |
|
|---|---|---|---|---|---|---|---|
single-core code |
22.5 s |
22.5 |
170 s |
170 s |
|||
1 |
29.6 s |
0.76 |
29.6 |
201 s |
.84 |
201 s |
|
2 |
17.7 s |
1.27 |
35.4 |
116 s |
1.5 |
232 s |
|
4 |
14.4 s |
1.56 |
57.6 |
82 s |
2.1 |
328 s |
|
8 |
12.1 s |
1.86 |
96.8 |
79 s |
2.2 |
632 s |
For word ngrams, it doesn’t even seem justifiable to use two processes because the speed up there is not even 1.5. In this case, the problem is that the code isn’t very efficient and is spending too much time passing the data around. Using an array job allows every array task to write separately, and then one single-core job is used to accumulate the counts. Better yet would be to re-do the code so that this inefficiency is improved.
From our experience, some of the main slow points of the code are:
Reading all the individual files (even from the zip file) is slow.
Writing the results in plain text + JSON is slow: using a binary format of some form would be better.
The python
multiprocessingmodule has to inefficiently move data between processes. Since this is a heavily data-based computation (and there are quite a few ngrams it makes), it is slow.
Shared memory parallelism 1: Test the example’s scaling
Run the example with a bigger number of trials (100000000 or \(10^{8}\))
and with 1, 2 and 4 CPUs. Check the running time and CPU
utilization for each run.
Solution
You can run the program without parallelization with:
srun --time=00:10:00 --mem=1G python3 slurm/pi.py 100000000
Afterwards you can use seff JOBID to get the utilization.
You can run the program with multiple CPUs with:
srun --cpus-per-task=2 --time=00:10:00 --mem=1G python3 slurm/pi.py --nprocs=2 100000000
srun --cpus-per-task=4 --time=00:10:00 --mem=1G python3 slurm/pi.py --nprocs=4 100000000
You should see that the time needed to run the program (“Job Wall-clock time”) ) is basically divided by the number of processors while the CPU utilization time (“CPU Utilized”) remains the same.
Shared memory parallelism 2: Test scaling for a program that has a serial part
pi.py can be called with an argument --serial=0.1 to run a
fraction of the trials in a serial fashion (here, 10%).
Run the example with a bigger number of trials (100000000 or \(10^{8}\)),
4 CPUs and a varying serial fraction (0.1, 0.5, 0.8). Check the running time and CPU
utilization for each run.
Solution
You can run the program with 10% serial execution using the following:
srun --cpus-per-task=4 --time=00:10:00 --mem=1G python3 slurm/pi.py --serial=0.1 --nprocs=4 100000000
Afterwards you can use seff JOBID to get the utilization.
Doing the run with different serial portion should show that a bigger the serial portion, the less benefit the parallelization gives.
Shared memory parallelism 3: More parallel \(\neq\) fastest solution
pi.py can be called with an argument --optimized to run an optimized
version of the code that utilizes NumPy
for vectorized calculations.
Run the example with a bigger number of trials (100000000 or \(10^{8}\)) and with
4 CPUs. Now run the optimized example with the same amount of trials and with 1 CPU.
Check the CPU utilization and running time for each run.
Solution
You can run the program with 4 CPUs using the following:
srun --cpus-per-task=4 --time=00:10:00 --mem=1G python3 slurm/pi.py --nprocs=4 100000000
You can run the optimized version with the following:
srun --time=00:10:00 --mem=1G python3 slurm/pi.py --optimized 100000000
Afterwards you can use seff JOBID to get the utilization.
The optimized version, which uses NumPy to create a big batch of random numbers at a time and calculates the hits for all of the random numbers at a same time should be significantly faster. NumPy itself uses libraries written in C and Fortran that make the calculations a lot faster than Python would.
Using libraries and coding practices that are better suited for the task can provide bigger performance boost that using multiple CPUs.
Shared memory parallelism 4: Your program
Think of your program. Do you think it can use shared-memory parallelism?
If you do not know, you can check the program’s documentation for words such as:
nprocs
nworkers
num_workers
njobs
OpenMP
…
These usually point towards some method of shared-memory parallel execution.
What’s next?
The next tutorial is about MPI parallelism.