Parallel computing: different methods explained
Videos
Videos of this topic may be available from one of our kickstart course playlists: 2023, 2022 Summer, 2022 February, 2021 Summer, 2021 February.
Parallel computing is what HPC is really all about: processing things on more than one processor at once. By now, you should have read all of the previous tutorials.
Abstract
You need to figure out what parallelization paradigm your program uses, otherwise you won’t know which options to use.
Embarrassingly parallel: use options for array jobs.
Multithreaded (OpenMP) or multiple processes (like Python’s multiprocessing): use options for shared memory parallelism.
MPI: use options for MPI parallelism.
GPU: use options for GPUs.
You must always monitor jobs to make sure they are using all the resources you request (
seff JOBID).If you aren’t fully sure of how to scale up, contact us Research Software Engineers early.
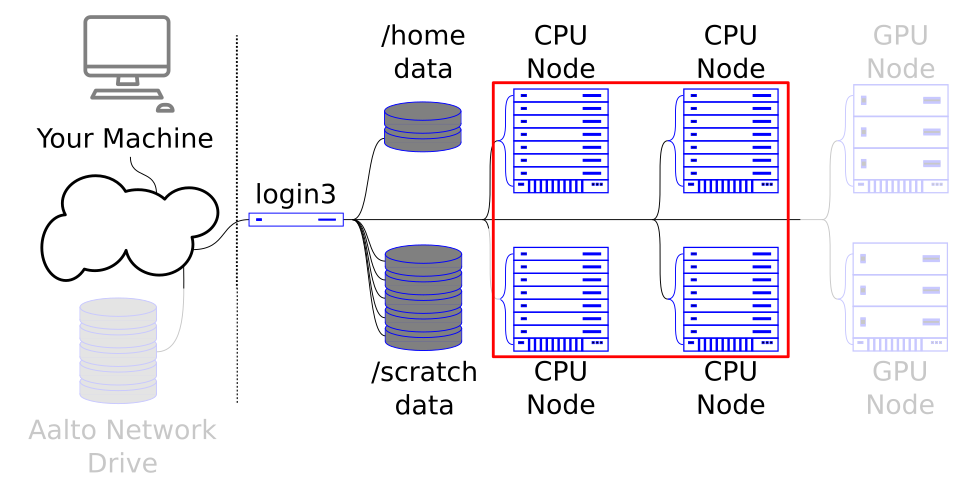
We are working to get access to the login node. This is the gateway to all the rest of the cluster.
Parallel programming models
Parallel programming is used to create programs that can execute instructions on multiple processors at a same time. Most of our users that run their programs in parallel utilize existing parallel execution features that are present in their programs and thus do not need to learn how to create parallel programs. But even when one is running programs in parallel, it is important to understand different models of parallel execution.
The main models of parallel programming are:
Embarrassingly parallel problem can be split into completely independent jobs that can be executed separately with no communication between individual jobs.
More often than not, scientific problems involve running a single program again and again with different datasets or parameters. Slurm has a structure called job array, which enables users to easily submit a large amount of such jobs.
Any program can be run in an embarassingly parallel way as long as the problem at hand can be split into multiple independent jobs.
Each job in an array is identical to every other job, but each independent job gets its own unique ID.
Workloads that utilize this model should request what a single job needs and the number of array jobs that the whole array should have.
See: array jobs.
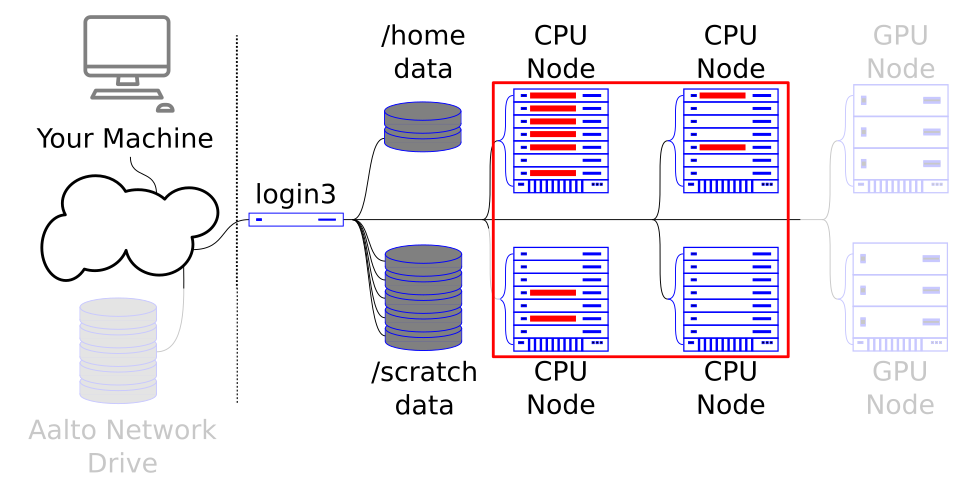
The array job runs independently across the cluster.

Shared memory (or multithreaded/multiprocess) parallel programs run multiple processes / threads on the same machine. As the name suggests, all of the computer’s memory has to be accessible to all of the processes / threads.
Thus programs that utilize this model should request one node, one task and multiple CPUs.
Example applications that utilize this model: Matlab (internally & parallel pool), R (internally & parallel-library), Python (numpy internally & threading/multiprocessing-modules), OpenMP applications, BLAS libraries, FFTW libraries, typical multithreaded/multiprocess parallel desktop programs.
See: shared-memory parallelism.
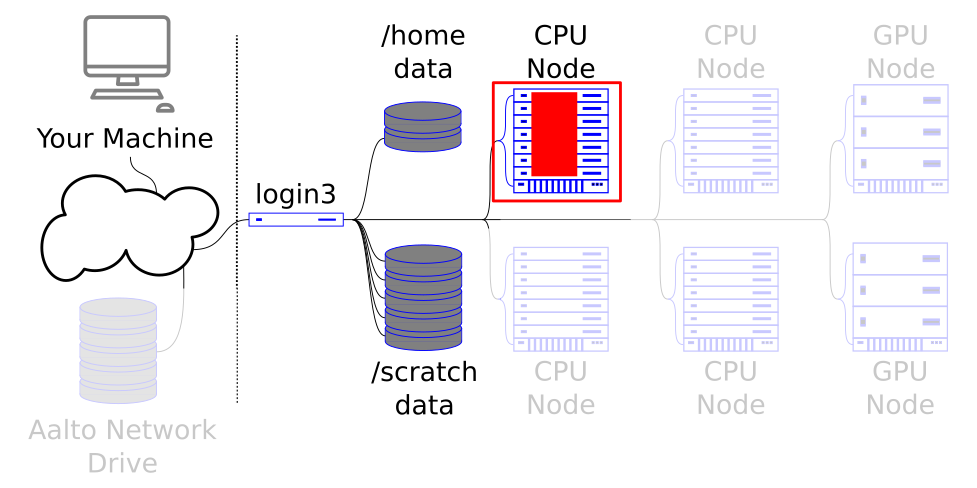
The shared memory job runs across one node - since that’s what shares memory.

MPI parallelism utilizes MPI (Message Passing Interface) libraries for communication between MPI tasks. These MPI tasks work in a collective fashion and each task executes its part of the same program.
Communication between MPI tasks is passed through the high-speed interconnects between different compute nodes and this allows for programs that can tuilize thousands of CPU cores.
Almost all large-scale scientific programs utilize MPI. MPI programs are usually quite complex and written for a specific use case as the nature of the collective operations depends on the problem at hand.
Programs that utilize this model should request single/multiple nodes with multiple tasks each. You should not request multiple CPUs per task.
Example applications that utilize this model: CP2K, GPAW, LAMMPS, OpenFoam. See: MPI parallelism.
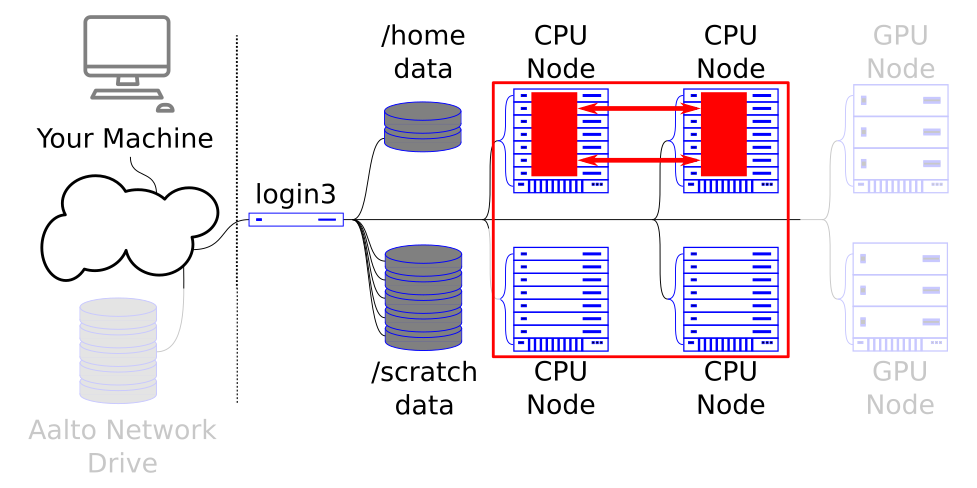
The MPI job can communicate across nodes.

Parallel execution in GPUs is not parallel in the traditional sense where multiple CPUs run different processes. Instead GPU parallelism leverages GPGPUs (general-purpose graphics processing units) that have thousands of compute cores inside them. When running suitable problems GPUs can be substantially faster than CPUs.
Programs that utilize GPUs are written in parts where some part of the program executes on the CPU and other is executed on the GPU. The part that runs on the CPU usually does things like reading input and writing output, while the GPU part is more focused on doing numerical calculations. Often multiple CPUs are needed per GPU to do things such as data preprocessing just to keep the GPU preoccupied.
A typical CPU program cannot utilize GPUs unless it has been designed to use them. Additionally programs that utilize GPUs cannot utilize multiple GPUs unless they have been designed for it.
Programs that utilize GPUs should request a single node, a single task, (optionally) multiple CPUs and a GPU.
See: GPU computing.

Does my code parallelize?
Normal serial code can’t just be run in parallel without modifications. As a user it is your responsibility to understand what parallel model implementation your code has, if any.
When deciding whether using parallel programming is worth the effort, one should be mindful of Amdahl’s law and Gustafson’s law. All programs have some parts that can only be executed in serial fashion and thus speedup that one can get from using parallel execution depends on how much of programs’ execution can be done in parallel.

Thus if your program runs mainly in serial but has a small parallel part, running it in parallel might not be worth it. Sometimes, doing data parallelism with e.g. array jobs is much more fruitful approach.
Another important note regarding parallelism is that all the applications scale good up to some upper limit which depends on application implementation, size and type of problem you solve and some other factors. The best practice is to benchmark your code on different number of CPU cores before you start actual production runs.
If you want to run some program in parallel, you have to know something about it - is it shared memory or MPI? A program doesn’t magically get faster when you ask more processors if it’s not designed to.
Combining different parallel execution models
Different parallel execution models can be combined if your program supports them. Below a few common situations are listed:
Embarassingly parallel everything
As running programs in an embarassingly parallel fashion is not a feature of the program, but a feature of the workflow itself, any program can be run in an embarassingly parallel fashion if needed.
One can run shared-memory parallel, MPI parallel and GPU parallel jobs in array jobs as well. Each individual job will get their own resources.
Hybrid parallelism
When MPI and shared memory parallelism are done by the same application it is usually called hybrid parallelization. Programs that utilize this model can require both multiple tasks and multiple CPUs per task.
For example, CP2K compiled to psmp-target has hybrid parallelization enabled
while popt-target has only MPI parallelization enabled. The best ratio between
MPI tasks and CPUs per tasks depends on the program and needs to be measured.
Multi-node parallelism without MPI
Some programs can run with multiple nodes in parallel, but they do not use MPI for communication between nodes. Resources for these programs are reserved in a similar fashion to the MPI programs, but the program launch is usually done by scripts that run different instructions on different machines. The setup depends on the program and can be complex.
See also
The Research Software Engineers can help in all aspects of parallel computing - we’d recommend anyone getting to this point set up a consultation to make sure your work is as efficient as it can be.
What’s next?
The next tutorial is about array jobs.