MPI parallelism: multi-task programs
Videos
Videos of this topic may be available from one of our kickstart course playlists: 2025 summer, 2025 winter, 2024, 2023, 2022 Summer, 2022 February, 2021 Summer, 2021 February. Or see computing explained with HPC kitchen.
Abstract
Verify that your program can use MPI.
Compile to link with our MPI libraries. Remember to load the same modules in your Slurm script.
Use
--nodes=1and--ntasks=nto reserve \(n\) tasks for your job.Start your application via
srunif using module installed MPI ormpirunif you have your own installation of MPI.For spreading tasks evenly across nodes, use
--nodes=Nand--ntasks-per-node=nfor getting \(N \cdot n\) tasks.You must always monitor jobs to make sure they are using all the resources you request (
seff JOBID).If you aren’t fully sure of how to scale up, contact us Research Software Engineers early.
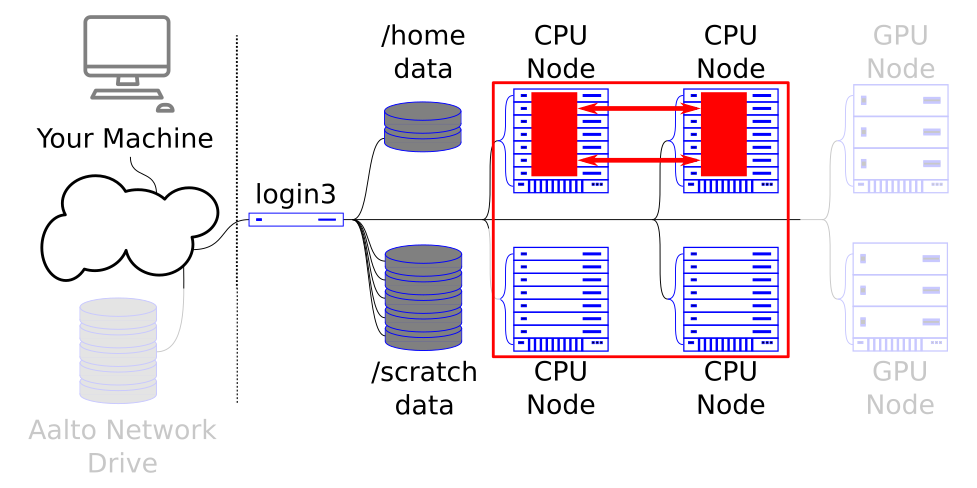
MPI parallelism lets you scale to many nodes on the cluster, at the cost of extra programming work.
What is MPI parallelism?
MPI or message passing interface is a standard for creating communication between many tasks that collectively run a program in parallel. Programs using MPI can scale up to thousands of nodes.
Programs using MPI need to be written so that they utilize the MPI communication. Thus typical programs that are not written around MPI cannot use MPI without major modifications.

MPI programs typically work in the following way:
Same program is started in multiple separate tasks.
All tasks join a communication layer with MPI.
Each tasks gets their own rank (basically and ID number).
Based on their ranks tasks execute their part of the code and communicate to other tasks. Rank 0 is usually the “main program” that prints output for monitoring.
After the program finishes communication layer is stopped.
When using module installed installations of MPI the MPI ranks will automatically get information on their ranks from Slurm via library called PMIx. If the MPI used is some other version, they might not connect with the Slurm correctly.
Running a typical MPI program
Compiling a MPI program
For compiling/running an MPI job one has to pick up one of the MPI library
suites. There are various different MPI libraries that all implement the
MPI standard. We recommend that you use our OpenMPI installation
(openmpi/4.1.6). For information on other installed versions, see the
MPI applications page.
Some libraries/programs might have already existing requirement for a certain MPI version. If so, use that version or ask for administrators to create a version of the library with dependency on the MPI version you require.
Warning
Different versions of MPI are not compatible with each other. Each version of MPI will create code that will run correctly with only that version of MPI. Thus if you create code with a certain version, you will need to load the same version of the library when you are running the code.
Also, the MPI libraries are usually linked to slurm and network drivers. Thus, when slurm or driver versions are updated, some older versions of MPI might break. If you’re still using said versions, let us know. If you’re just starting a new project, it is recommended to use our recommended MPI libraries.
Reserving resources for MPI programs
For basic use of MPI programs, you will need to use the
--nodes=1 and --ntasks=N-options to specify the number of MPI workers.
The --nodes=1 option is recommended so that your jobs will run in the
same machine for maximum communication efficiency. You can also run
without it, but this can result in worse performance.
In many cases you might require more tasks than one node has CPUs.
When this is the case, it is recommended to split the number of
workers evenly among the nodes. To do this, one can use
--nodes=N and --ntasks-per-node=n. This would give you
\(N \cdot n\) tasks in total.
Each task will get a default of 1 CPU. See section on hybrid parallelisation for information on whether you can give each task more than one CPU.
Running and example MPI program
The scripts you need for the following exercises can be found in our
hpc-examples, which
we discussed in Using the cluster from a command line (section
Copy your code to the cluster).
You can clone the repository by running
git clone https://github.com/AaltoSciComp/hpc-examples.git. Doing this
creates you a local copy of the repository in your current working
directory. This repository will be used for most of the tutorial exercises.
For this example, let’s consider
pi-mpi.c-example
in the slurm-folder.
It estimates pi with Monte Carlo methods and
can utilize multiple MPI tasks for calculating the trials.
First off, we need to compile the program with a suitable OpenMPI version. Let’s use the
recommended version openmpi/4.1.6:
$ module load triton/2024.1-gcc openmpi/4.1.6
$ mpicc -o pi-mpi slurm/pi-mpi.c
The program can now be run with srun ./pi-mpi N, where N is the number of
iterations to be done by the algorithm.
Let’s ask for resources and run the program with two processes using srun:
$ srun --nodes=1 --ntasks=2 --time=00:10:00 --mem=500M ./pi-mpi 1000000
This worked because we had the correct modules already loaded. Using a slurm script setting the requirements and loading the correct modules becomes easier:
#!/bin/bash
#SBATCH --time=00:10:00
#SBATCH --mem=500M
#SBATCH --output=pi-mpi.out
#SBATCH --nodes=1
#SBATCH --ntasks=2
module load triton/2024.1-gcc openmpi/4.1.6
srun ./pi-mpi 1000000
Let’s call this script pi-mpi.sh. You can submit it with:
$ sbatch pi-mpi.sh
Special cases and common pitfalls
MPI workers do not see each other
When using our installations of MPI the MPI ranks will automatically get information on their ranks from Slurm via library called PMIx. If the MPI used is some other version, they might not connect with the Slurm correctly.
If you have your own installation of MPI you might try setting
export SLURM_MPI_TYPE=pmix_v2 in your job before calling srun.
This will tell Slurm to use PMIx for connecting with the MPI installation.
Setting a constraint for a specific CPU architecture
The number of CPUs/tasks one can specify for a single parallel job depends usually on the underlying algorithm. In many codes, such as many finite-difference codes, the workers are set in a grid-like structure. The user of said codes has then a choice of choosing the dimensions of the simulation grid aka. how many workers are in x-, y-, and z-dimensions.
For best perfomance one should reserve half or full nodes when possible. In heterogeneous clusters this can be a bit more complicated as different CPUs can have different numbers of cores.
In Triton CPU partitions there are machines with 24, 28 and 40 CPUs. See the list of available nodes for more information.
However, one can make the reservations easier by specifying a CPU architecture
with --constraint=ARCHITECTURE. This tells Slurm to look for nodes that
satisfy a specific feature. To list available features, one can use
slurm features.
For example, one could limit the code to the Haswell-architecture with the following script:
#!/bin/bash
#SBATCH --time=00:10:00 # takes 5 minutes all together
#SBATCH --mem-per-cpu=200M # 200MB per process
#SBATCH --nodes=1 # 1 node
#SBATCH --ntasks-per-node=24 # 24 processes as that is the number in the machine
#SBATCH --constraint=hsw # set constraint for processor architecture
module load triton/2024.1-gcc openmpi/4.1.6 # NOTE: should be the same as you used to compile the code
srun ./pi-mpi 1000000
Monitoring performance
You can use seff JOBID to see what percent of available CPUs and RAM was
utilized. Example output is given below:
$ seff 60985042
Job ID: 60985042
Cluster: triton
User/Group: tuomiss1/tuomiss1
State: COMPLETED (exit code 0)
Nodes: 1
Cores per node: 2
CPU Utilized: 00:00:29
CPU Efficiency: 90.62% of 00:00:32 core-walltime
Job Wall-clock time: 00:00:16
Memory Utilized: 1.59 MB
Memory Efficiency: 0.08% of 2.00 GB
If your processor usage is far below 100%, your code may not be working correctly. If your memory usage is far below 100% or above 100%, you might have a problem with your RAM requirements. You should set the RAM limit to be a bit above the RAM that you have utilized.
You can also monitor individual job steps by calling seff with the syntax
seff JOBID.JOBSTEP.
Important
When making job reservations it is important to distinguish
between requirements for the whole job (such as --mem) and
requirements for each individual task/cpu (such as --mem-per-cpu).
E.g. requesting --mem-per-cpu=2G with --ntasks=2 and --cpus-per-task=4
will create a total memory reservation of
(2 tasks)*(4 cpus / task)*(2GB / cpu)=16GB.
Hybrid parallelization aka. giving more than one CPU to each MPI task
When MPI and shared memory parallelism are done by the same application it is usually called hybrid parallelization. Programs that utilize this model can require both multiple tasks and multiple CPUs per task.
For example, CP2K compiled to psmp-target has hybrid parallelization enabled
while popt-target has only MPI parallelization enabled. The best ratio between
MPI tasks and CPUs per tasks depends on the program and needs to be measured.
Remember that the number of CPUs in a machine is hardware dependent.
The total number of CPUs per node when you request --ntasks-per-node=n and
--cpus-per-task=C is \(n \cdot C\). This number needs to be equal or less than
the total number of CPUs in the machine.
Exercises
The scripts you need for the following exercises can be found in our
hpc-examples, which
we discussed in Using the cluster from a command line (section
Copy your code to the cluster).
You can clone the repository by running
git clone https://github.com/AaltoSciComp/hpc-examples.git. Doing this
creates you a local copy of the repository in your current working
directory. This repository will be used for most of the tutorial exercises.
MPI parallelism 3: Your program
Think of your program. Do you think it can use MPI parallelism?
If you do not know, you can check the program’s documentation for words such as:
MPI
message-passing interface
mpirun
mpiexec
…
These usually point towards some method of MPI parallel execution.
What’s next?
The next tutorial is about GPU parallelism.