GPU computing
Videos
Videos of this topic may be available from one of our kickstart course playlists: 2023, 2022 Summer, 2022 February, 2021 Summer, 2021 February.
Abstract
Request a GPU with the Slurm option
--gres=gpu:1or--gpus=1(some clusters need-p gpuor similar).Select a certain type of GPU with e.g.
--constraint='volta'(see the quick reference for names).Monitor GPU performance with
sacct -j JOBID -o comment -p.For development, run jobs of 4 hours or less, and they can run quickly in the
gpushortqueue.If you aren’t fully sure of how to scale up, contact us Research Software Engineers early.
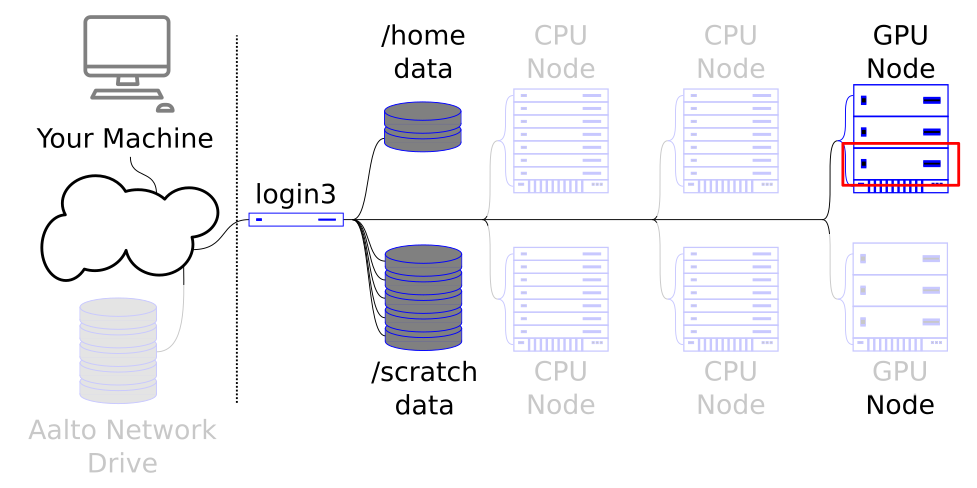
GPU nodes allow specialized types of work to be done massively in parallel.
What are GPUs and how do they parallelise calculations?
GPUs, short for graphical processing unit, are massively-parallel processors that are optimized to perform numerical calculations in parallel. Due to this specialisation GPUs can be substantially faster than CPUs when solving suitable problems.
GPUs are especially handy when dealing with matrices and vectors. This has allowed GPUs to become an indispensable tool in many research fields such as deep learning, where most of the calculations involve matrices.
The programs we normally write in common programming languages, e.g. C++ are executed by the CPU. To run a part of that program in a GPU the program must do the following:
Specify a piece of code called a kernel, which contains the GPU part of the program and is compiled for the specific GPU architecture in use.
Transfer the data needed by the program from the RAM to GPU VRAM.
Execute the kernel on the GPU.
Transfer the results from GPU VRAM to RAM.

To help with this procedure special APIs (application programming interfaces) have been created. An example of such an API is CUDA toolkit, which is the native programming interface for NVIDIA GPUs.
On Triton, we have a large number of NVIDIA GPU cards from different generations and a single machine with AMD GPU cards. Triton GPUs are not the typical desktop GPUs, but specialized research-grade server GPUs with large memory, high bandwidth and specialized instructions. For scientific purposes, they generally outperform the best desktop GPUs.
See also
Please ensure you have read Interactive jobs and Serial Jobs before you proceed with this tutorial.
Running a typical GPU program
Reserving resources for GPU programs
Slurm keeps track of the GPU resources as generic resources (GRES) or trackable resources (TRES). They are basically limited resources that you can request in addition to normal resources such as CPUs and RAM.
To request GPUs on Slurm, you should use the --gres=gpu:1 or --gpus=1
-flags.
You can also use syntax --gres=gpu:GPU_TYPE:1, where GPU_TYPE
is a name chosen by the admins for the GPU. For example, --gres=gpu:v100:1
would give you a V100 card. See section on
reserving specific GPU architectures for more information.
You can request more than one GPU with --gres=gpu:G, where G is
the number of the requested GPUs.
Some GPUs are placed in a quick debugging queue. See section on reserving quick debugging resources for more information.
Note
Most GPU programs cannot utilize more than one GPU at a time. Before trying to reserve multiple GPUs you should verify that your code can utilize them.
Running an example program that utilizes GPU
The scripts you need for the following exercises can be found in our
hpc-examples, which
we discussed in Using the cluster from a shell.
You can clone the repository by running
git clone https://github.com/AaltoSciComp/hpc-examples.git. Doing this
creates you a local copy of the repository in your current working
directory. This repository will be used for most of the tutorial exercises.
For this example, let’s consider
pi-gpu.cu
in the slurm-folder.
It estimates pi with Monte Carlo methods and can utilize a GPU for calculating
the trials.
The script is in the slurm-folder. This example is written in C++ and CUDA.
Thus it needs to be compiled before it can be run.
To compile CUDA-based code for GPUs, lets load a cuda-module and
a newer compiler:
module load gcc/8.4.0 cuda
Now we should have a compiler and a CUDA toolkit loaded. After this we can compile the code with:
nvcc -arch=sm_60 -gencode=arch=compute_60,code=sm_60 -gencode=arch=compute_70,code=sm_70 -gencode=arch=compute_80,code=sm_80 -o pi-gpu pi-gpu.cu
This monstrosity of a command is written like this because we want our code to be able run on multiple different GPU architectures. For more information, see section on setting compilation flags for GPU architectures.
Now we can run the program using srun:
srun --time=00:10:00 --mem=500M --gres=gpu:1 ./pi-gpu 1000000
This worked because we had the correct modules already loaded. Using a slurm script setting the requirements and loading the correct modules becomes easier:
#!/bin/bash
#SBATCH --time=00:10:00
#SBATCH --mem=500M
#SBATCH --output=pi-gpu.out
#SBATCH --gres=gpu:1
module load gcc/8.4.0 cuda
./pi-gpu 1000000
Note
If you encounter problems with CUDA libraries, see the section on missing CUDA libraries.
Special cases and common pitfalls
Monitoring efficient use of GPUs
When running a GPU job, you should check that the GPU is being fully utilized.
When your job has started, you can ssh to the node and run
nvidia-smi. It should be close to 100%.
Once the job has finished, you can use slurm history to obtain the
jobID and run:
$ sacct -j JOBID -o comment -p
{"gpu_util": 99.0, "gpu_mem_max": 1279.0, "gpu_power": 204.26, "ncpu": 1, "ngpu": 1}|
This also shows the GPU utilization.
If the GPU utilization of your job is low, you should check whether
its CPU utilization is close to 100% with seff JOBID. Having a high
CPU utilization and a low GPU utilization can indicate that the CPUs are
trying to keep the GPU occupied with calculations, but the workload
is too much for the CPUs and thus GPUs are not constantly working.
Increasing the number of CPUs you request can help, especially in tasks that involve data loading or preprocessing, but your program must know how to utilize the CPUs.
However, you shouldn’t request too many CPUs: There wouldn’t be enough CPUs for everyone to use the GPUs and they would go to waste (all of our nodes have 4-12 CPUs for each GPU).
Reserving specific GPU types
You can restrict yourself to a certain type of GPU card by using
using the --constraint option. For example, to restrict the submission to
Pascal generation GPUs only you can use --constraint='pascal'.
For choosing between multiple generations, you can use the |-character
between generations. For example, if you want to restrict the submission
Volta or Ampere generations you can use --constraint='volta|ampere'.
Remember to use the quotes since | is the shell pipe.
To see what GPU resources are available, run slurm features or
sinfo -o '%50N %18F %26f %30G'.
Alternative way is to use syntax --gres=gpu:GPU_TYPE:1, where GPU_TYPE
is a name chosen by the admins for the GPU. For example, --gres=gpu:v100:1
would give you a V100 card.
Reserving resources from the short job queue for quick debugging
There is a gpushort partition with a time limit of 4 hours that
often has space (like with other partitions, this is automatically
selected for short jobs). As of early 2022, it has four Tesla P100
cards in it (view with slurm partitions | grep gpushort). If you
are doing testing and development and these GPUs meet your needs, you
may be able to test much faster here. Use -p gpushort for this.
CUDA libraries not found
If you ever get libcuda.so.1: cannot open shared object file: No such
file or directory, this means you are attempting to use a CUDA
program on a node without a GPU. This especially happens if you try
to test a GPU code on the login node.
Another problem that might occur is when a program will try to use pre-compiled kernels, but the corresponding CUDA toolkit is not available.
This might happen in you have used a cuda-module to compile
the code and it is not loaded when you try to run the code.
If you’re using Python, see the section on CUDA libraries and Python.
CUDA libraries and Python deep learning frameworks
When using a Python deep learning frameworks such as Tensorflow or PyTorch you usually need to create a conda environment that contains both the framework and CUDA framework that the framework needs.
We recommend that you either use our centrally installed module that contains both frameworks (more info here) or install your own using environment using instructions presented here. These instructions make certain that the installed framework has a corresponding CUDA toolkit available. See the application list for more details on specific frameworks.
Please note that pre-installed software either has CUDA already present or it loads the needed modules. Thus you do not need to explicitly load CUDA from the module system when loading these.
Setting CUDA architecture flags when compiling GPU codes
Many GPU codes come with precompiled kernels, but in some cases you might need to compile your own kernels. When this is the case you’ll want to give the compiler flags that make it possible to run the code on multiple different GPU architectures.
For GPUs in Triton these flags are:
-arch=sm_60 -gencode=arch=compute_60,code=sm_60 -gencode=arch=compute_70,code=sm_70 -gencode=arch=compute_80,code=sm_80
Here architectures (compute_XX/sm_XX) number 60, 70 and 80
correspond to GPU cards P100, V100 and A100 respectively.
For more information, you can check this excellent article or CUDA documentation on the subject.
Keeping GPUs occupied when doing deep learning
Many problems such as deep learning training are data-hungry. If you are loading large amounts of data you should make certain that the data loading is done in an efficient manner or the GPU will not be fully utilized.
All deep learning frameworks have their own guides on how to optimize the data loading, but they all are some variation of:
Store your data in multiple big files.
Create code that loads data from these big files.
Run optional pre-processing functions on the data.
Create a batch of data out of individual data samples.
Tasks 2 and 3 are usually parallelized across multiple CPUs. Using pipelines such as these can dramatically speed up the training procedure.
If your data consists of individual files that are not too big,
it is a good idea to have the data stored in one file, which is then
copied to nodes ramdisk /dev/shm or temporary disk /tmp.
Avoiding small files is in general a good rule to follow. Please refer to the small files page for more detailed information.
If your data is too big to fit in the disk, we recommend that you contact us for efficient data handling models.
For more information on suggested data loading procedures for different frameworks, see Tensorflow’s and PyTorch’s guides on efficient data loading.
Profiling GPU usage with nvprof
When using NVIDIA’s GPUs you can try to use a profiling tool
called nvprof to monitor what took most of the GPU’s
time during the code’s execution.
Sample output might look something like this:
==30251== NVPROF is profiling process 30251, command: ./pi-gpu 1000000000
==30251== Profiling application: ./pi-gpu 1000000000
==30251== Profiling result:
Type Time(%) Time Calls Avg Min Max Name
GPU activities: 84.82% 11.442ms 1 11.442ms 11.442ms 11.442ms throw_dart(curandStateXORWOW*, int*, unsigned long*)
14.70% 1.9833ms 1 1.9833ms 1.9833ms 1.9833ms setup_rng(curandStateXORWOW*, unsigned long)
0.30% 40.704us 1 40.704us 40.704us 40.704us [CUDA memcpy DtoH]
0.17% 23.328us 1 23.328us 23.328us 23.328us [CUDA memcpy HtoD]
API calls: 89.52% 122.81ms 3 40.936ms 3.6360us 122.70ms cudaMalloc
10.05% 13.794ms 2 6.8969ms 68.246us 13.726ms cudaMemcpy
0.20% 269.55us 3 89.851us 11.283us 130.45us cudaFree
0.14% 196.08us 101 1.9410us 122ns 83.854us cuDeviceGetAttribute
0.04% 57.228us 2 28.614us 6.3760us 50.852us cudaLaunchKernel
0.02% 32.426us 1 32.426us 32.426us 32.426us cuDeviceGetName
0.01% 13.677us 1 13.677us 13.677us 13.677us cuDeviceGetPCIBusId
0.01% 10.998us 1 10.998us 10.998us 10.998us cudaGetDevice
0.00% 2.3540us 1 2.3540us 2.3540us 2.3540us cudaGetDeviceCount
0.00% 1.2690us 3 423ns 207ns 850ns cuDeviceGetCount
0.00% 663ns 2 331ns 170ns 493ns cuDeviceGet
0.00% 656ns 1 656ns 656ns 656ns cuDeviceTotalMem
0.00% 396ns 1 396ns 396ns 396ns cuModuleGetLoadingMode
0.00% 234ns 1 234ns 234ns 234ns cuDeviceGetUuid
This output shows that most of the computing time was caused by calling the
throw_dart-kernel. It is important to note that in this example memory
allocation cudaMalloc and memory copying cudaMemcpy used more time than
the actual computation. Memory operations are time consuming operations and thus
best codes try to minimize the need for doing them.
To see a chronological order of different GPU operations one can also run
nprof --print-gpu-trace. The output will look something like this:
==31050== NVPROF is profiling process 31050, command: ./pi-gpu 1000000000
==31050== Profiling application: ./pi-gpu 1000000000
==31050== Profiling result:
Start Duration Grid Size Block Size Regs* SSMem* DSMem* Size Throughput SrcMemType DstMemType Device Context Stream Name
182.84ms 23.136us - - - - - 256.00KB 10.552GB/s Pageable Device Tesla P100-PCIE 1 7 [CUDA memcpy HtoD]
182.89ms 1.9769ms (512 1 1) (128 1 1) 31 0B 0B - - - - Tesla P100-PCIE 1 7 setup_rng(curandStateXORWOW*, unsigned long) [118]
184.87ms 11.450ms (512 1 1) (128 1 1) 19 0B 0B - - - - Tesla P100-PCIE 1 7 throw_dart(curandStateXORWOW*, int*, unsigned long*) [119]
196.33ms 40.704us - - - - - 512.00KB 11.996GB/s Device Pageable Tesla P100-PCIE 1 7 [CUDA memcpy DtoH]
Regs: Number of registers used per CUDA thread. This number includes registers used internally by the CUDA driver and/or tools and can be more than what the compiler shows.
SSMem: Static shared memory allocated per CUDA block.
DSMem: Dynamic shared memory allocated per CUDA block.
SrcMemType: The type of source memory accessed by memory operation/copy
DstMemType: The type of destination memory accessed by memory operation/copy
Here we see that the sample code did a memory copy to the device, ran kernel setup_rng,
ran kernel throw_dart and did a memory copy back to the host memory.
For more information on nvprof, see NVIDIA’s documentation on it.
Available GPUs and architectures
Card |
Slurm feature name ( |
Slurm gres name ( |
total amount |
nodes |
architecture |
compute threads per GPU |
memory per card |
CUDA compute capability |
|---|---|---|---|---|---|---|---|---|
Tesla K80* |
|
|
12 |
gpu[20-22] |
Kepler |
2x2496 |
2x12GB |
3.7 |
Tesla P100 |
|
|
20 |
gpu[23-27] |
Pascal |
3854 |
16GB |
6.0 |
Tesla V100 |
|
|
40 |
gpu[1-10] |
Volta |
5120 |
32GB |
7.0 |
Tesla V100 |
|
|
40 |
gpu[28-37] |
Volta |
5120 |
32GB |
7.0 |
Tesla V100 |
|
|
16 |
dgx[1-7] |
Volta |
5120 |
16GB |
7.0 |
Tesla A100 |
|
|
56 |
gpu[11-17,38-44] |
Ampere |
7936 |
80GB |
8.0 |
AMD MI100 (testing) |
|
Use |
gpuamd[1] |
Exercises
The scripts you need for the following exercises can be found in our
hpc-examples, which
we discussed in Using the cluster from a shell.
You can clone the repository by running
git clone https://github.com/AaltoSciComp/hpc-examples.git. Doing this
creates you a local copy of the repository in your current working
directory. This repository will be used for most of the tutorial exercises.
GPU 1: Test nvidia-smi
Run nvidia-smi on a GPU node with srun. Use slurm history
to check which GPU node you ended up on.
GPU 2: Running the example
Run the example given above with larger number of trials
(10000000000 or \(10^{10}\)).
Try using sbatch and Slurm script as well.
GPU 3: Run the script and do basic profiling with nvprof
nvprof is part of NVIDIA’s profiling tools and it can be
used to monitor which parts of the GPU code use up most time.
Run the program as before, but add nvprof before it.
Try running the program with chronological trace mode
(nvprof --print-gpu-trace) as well.
Solution
With srun you can run the profiling as follows:
srun --time=00:10:00 --mem=500M --gres=gpu:1 nvprof ./pi-gpu 10000000000
To get the trace output, you need to add the --print-gpu-trace-flag:
srun --time=00:10:00 --mem=500M --gres=gpu:1 nvprof --print-gpu-trace ./pi-gpu 10000000000
You should see output similar to ones shown in the section profiling GPU usage with nvprof.
GPU 4: Your program
Think of your program. Do you think it can utilize GPUs?
If you do not know, you can check the program’s documentation for words such as:
GPU
CUDA
ROCm
OpenMP offloading
OpenACC
OpenCL
…
See also
If you aren’t fully sure of how to scale up, contact us Research Software Engineers early.
What’s next?
You have now seen the basics - but applying these in practice is still a difficult challenge! There is plenty to figure out while combining your own software, the Linux environment, and Slurm.
Your time is the most valuable thing you have. If you aren’t fully sure of how to use the tools, it is much better to ask that struggle forever. Contact us the Research Software Engineers early - for example in our daily garage, and we can help you get set up well. Then, you can continue your learning while your projects are progressing.